Software in Medical Devices – Update for Q1/Q2 2025 The past year, as in previous…

Software in Medical Devices – Update for Q1/Q2 2019
Software in Medical Devices – Update for Q1/Q2 2019
This is a continuation of the software updates I have been sending out. Please check out all the references to download and/or to purchase. If you have any questions, please contact us.
Software is everywhere in medical devices and IVDs. The FDA and CE are becoming more pedantic on how they review and relate to software. The number of companies getting into the field is growing and the amount of software being developed for medical is very large.
The is an emphasis on “digital health” where the FDA is fast-tracking many devices (even though it is only software, it is still a medical device). Just because it is software only, this doesn’t mean that you are free from all the regulations, including a quality management system, risk analysis, etc.
There are rumors that the FDA will get rid of the differences in the documentation to submit for the Level Of Concern (LOC). If this happens, all submissions will probably be like a Major LOC of today, including the static code analysis report.
Software Recalls Q1-Q2 /2019
We have been following the recalls and there were a growing number of recalls that are listed where software played a role in the recall. It is interesting to note that software is the leading cause of recalls in the FDA for the past 3 years. This trend does not look like it will change.
The following are additional examples of recalls involving software directly as listed on the FDA website. There were over 150 recalls in this period relating to software, including several class 1 recalls. There may be more but classified not under software. The descriptions given for the recall are taken from the FDA database. For further details on the recalls, you can check them out on the FDA’s recall database.
- UniCel DxH 800 Coulter Cellular Analysis System, Class I – Complaints received of sporadic erroneously elevated platelet results without flags or system messages. Thrombocytopenia may go unrecognized.
- Spine & Trauma 3D Navigation 1.0, Class I – In certain occurrences, the affected navigation software application might unexpectedly display a navigated instrument in an axial, coronal/sagittal (ACS) view representation with fixed planes in the image reconstruction (the not updated ACS view ), instead of displaying the desirable view representation Inline View , which is commonly used for navigating invasive instruments at the spine. This could lead the surgeon to be unable to determine the position of the navigated instrument. This might occur after a crash restore or after changing between different navigation workflows during the same patient treatment.
- LIFEPAK 15 Monitor/Defibrillator, Class I – Certain LIFEPAK 15 Monitors/Defibrillators were reported to experience a lockup condition after a shock was delivered. This condition is defined as a blank monitor display with LED
lights on, indicating power on the device, but no response in keypad and device functions. - McKesson Cardiology Hemo, Class II – Users are not notified of procedure medication discrepancies between the Vitals and Meds, the Procedure Notes and Patient Common Data screens in Horizon/McKesson Cardiology Hemo” (Hemo). The discrepancy may also affect administered medication data in reports generated from Hemo or the Horizon/McKesson Cardiology Physician s Report as well as customers, who have implemented an outbound interface of procedure results.
- Stryker Sustainability Solutions (SSS) Reprocessed Arthroscopic Shavers, Class II – Affected lots of reprocessed arthroscopic shavers may display the incorrect catalog number and speed settings when connected to the generator, require manual mode for use on CORE generators, or display an error message on Crossfire 1 and 2 generators which does not allow the device to be used.
- VITEK 2 AST-N351 Test Kit, Class II – False Positive ESBL Phenotype.
- Anti-HAV IgM test System, Class II – Potential for erroneous result messages for the Anti-HAV IgM assay when run on the Cobas e 602 module. Discrepant result reporting may result.
- Kaluza C Flow Cytometry Software, Class II – Compatibility issue between the software and Microsoft updates to Windows 7, 8, and 10. The compatibility issue causes the software to be unusable which could result in a delay of reporting results.
- AMSCO 3000 Series Washer/Disinfector Model # 3052 and Model # 5052, Class II – The software in the systems may not process the cycle originally intended. This could result in damage to the various medical devices and instruments processed in the washer/disinfector or potential for inadequate cleaning or disinfection.
- MEVION S250i, Class II – Treatment is allowed to continue (via partials) in situations where dDose1 interlock has tripped.
- Monarch Medical Technologies EndoTool IV, Class II – Insulin dosing calculations were erroneously high.
- Prometra Programmer (Grand Prime) Software, Class II – The pump shuts down unexpectedly if Error 115, an alarm associated with a watchdog timer timeout, is triggered. There has been an increase of Error 115 occurrences due to a software defect caused by a Healthcare Provider using a Clinician Programmer, Software Version 2.00.29, to program a Bridge or Demand bolus while the pump is set to either a Periodic Flow or Multiple Rates flow mode.
- Biomerieux Vitek 2 Test kit, Class II – Unexpected ESBL (Extended Spectrum Beta Lactamase) phenotype had been proposed for some Escherichia coli strains in conjunction with the VITEK 2 test kits.
- MOSAIQ Oncology Information System and Sequencer, Class II – There is a potential that Wedge IDs were not included in the DICOM RT PLAN sent from the TPS, which will cause the field in MOSAIQ to be created with no wedge. Treatment of a field without the planned wedge will receive more dose than the treatment plan indicates.
- Elekta Unity, Image-Guided Radiation Therapy System, Class II – The QA software solution to perform the MR to MV alignment check, does not display the stored MR to MV offset values. The user is unable to independently inspect the values during their QA.
- HAMILTON-G5, Class II – New software version for affected ventilators reduces the probability of the ventilator entering an ambient state, in which the inspiratory channel and expiratory valves are opened, letting the patient breathe room air unassisted. When the ventilator enters the ambient state, alternative ventilation must be provided immediately.
- Radrex-i X-Ray System, Class II – software malfunction; It was found when a user performs radiography using the wireless flat panel detector (FPD), a message window displays on the monitor stating image transmission was not completed and there was no image available. It also showed the “OK” button to reacquire image data from the FPD, and the “Cancel” button to cancel the acquisition. When the user selects the “OK” button, the same message window appears. This prompted the user to repeat the same operation several times and finally select the “Cancel” button to quit the re-acquisition mode.
- RayStation x.y, Class II – When calculating electron Monte Carlo dose with a very large number of histories, the dose calculation may be wrong.
- SOMATOM go.Now, Class II – The potential sporadic performance problems may result in scanning workflow interruptions and unexpected user notifications. This may cause a delay in diagnosis or patient scans.
- EVOLIS Microplate System, Class II – User-induced circumstances can contribute to the EVOLIS Microplate Processor System not handling repeat testing requests/results properly and may contribute to transmitting erroneous results to the customer’s Laboratory Information System (LIS).
- PROBEAT-V, Class II – There is a potential for a discrepant target position when using 3D3D matching mode in PIAS (Positioning Image Analysis System) software installed in the PROBEAT-V, proton therapy systems.
- Centricity PACS-IW with Universal Viewer version 5.0, Class II – There is a potential that one or more images or image series may be missing from exams without a warning displayed in the viewer.
- AUTION HYBRID AU-4050, Class II – This correction is being initiated due to a software issue which results in the possibility of incorrect patient information being assigned to sample results when the following rare combination of three specific events were to occur: 1) No measurement results are generated due to an error by a urine sediment measurement. 2) The instrument is shutdown incorrectly. 3) An item rack is used for subsequent sample measurements.
- LVivo EF app on Vscan Extend, Class II – Overestimation bias in automatically calculated ejection fraction (EF) values while using LVivo EF app on the Vscan Extend product.
- GE Centricity PACS-IW, Class II – There is a potential that one or more image series (i.e., all images within an image set) may be missing from an exam without a warning displayed in the viewer.
- Philips IntelliBridge System, Class III – Infusion Pump Data Storage Accuracy-Data from the BBraun Space LAN or Arcomed UniqueDoc infusion pumps transmitted via the HL7 output interface through the Patient Information Center iX using a LAN driver may be recorded in the patients chart or electronic medical record at exactly 100 times the actual bolus rate, infusion rate and total volume values.
- InterStim System, Class II – There is a potential for an unexpected increase in stimulation during InterStim programming with the A10 Clinician Application (on Medtronic’s smart programmer).
- TactiSys Quartz Equipment, Class II – In reported cases, the device log on the TactiSys Quartz Equipment operating on Software Version 1.7.0 fills the allocated disk space, which prevents the storage of new log data. This may lead to intermittent contact force data to be displayed during the procedure.
- Leksell GammaPlan 11.1, Class II – The margin tool in Leksell GammaPlan 11.1 systematically overestimates margin in certain areas of the volume.
- NeuViz 128 Multi-slice CT Scanner System, Class II – Software defect: For Helical scan with ClearView function, when small arc (uninterrupted scan) occurs during scan, there will be some electromagnetic interference in the system which may affect the control signal of the flying focal spot, resulting in an incorrect flying focal spot sequence. If the system does not receive error, it would continue to complete the scan with the incorrect flying focal spot sequence, having an effect on the reconstruction of some images, resulting in image loss.
- VITROS XT 7600 Integrated System, Class II – Potential for sample fluid to be dispensed to an incorrect position on the MicroSlide, potentially leading to erroneous assay results being reported.
- Cobas Infinity Central Lab IT Solution, Class II – Using the following versions of cobas infinity software (2.0 thorough 2.5), there is a potential where the results from the first sample subsequent to the bottle change, (when using the Standby bottle function), does not display a quality control (QC) alarm when one should be present. If the QC is out of range on the stand-by bottle and it is not removed from the system, there is a potential that when the status of the Standby bottle changes from the Standby bottle to the Current bottle status, QC alarms will not be displayed next to the sample result.
- Artis Q, Class II – In affected Artis systems the movement of the floating tabletop may be blocked after a collision sensor has been activated during system movement.
- SureSigns VS3 NBP, Class II – System software inhibits the monitor and as a result does not measure, display and alarm for pulse rates above 240 beats per minute.
- VITROS 3600 Immunoassay System Refurbished, Class II – Luminometer Malfunction May Cause Inability to Process MicroWell Assays on VITROS Systems.
- SOMATOM Definition Edge, Class II – Potential risk of scans being aborted when using the optional Dual Spiral Dual Engery mode due to a software issue found in syngo CT VB10A for the Siemens Healthineers CT scanners with a single X-ray tube.
- Guardian Connect App CSS7200 used on the iPhone, iPad, and iPod Touch devices, Class II – The application may be closed by the operating system without alerting the user the app is no longer running or communicating with the transmitter resulting in the user not receiving alerts that could be associated with hypoglycemic or hyperglycemic events.
- MRIdian Linac Radiation Therapy System, Class II – A discrepancy between optimization and planning forward dose calculation between adaptive optimizations and AQA dose calculations can occur.
- St. Jude Medical Confirm Rx Insertable Cardiac Monitor, Class II – The device is unable to pair with the mobile app due to the device incorrectly determining the certificate has expired.
- VITROS XT 7600 Integrated System, Class II – One of the software algorithms used to detect sample dispense errors was inadvertently disabled. Because of this, sample dispense errors may lead to incorrect results being reported without an error code to alert the user.
- OnSight 3D Extremity System- X-Ray, Tomography Computed System, Class II – When the user performs the re-assignment of a parent / companion pair, the parent volume is transferred to the new patient, but the companion volume will remain in the original patient s exam.
- Olympus Diego Elite Console, Class II – Olympus Diego Elite Consoles may inadvertently permit activation of the RF energy feature when used with Diego Elite blades.
- Syngo.via syngo.CT Cardiac Function, Class II – There is a potential risk of a wrong measurement in the annulus plane during a TAVI planning procedure. This risk is due to a software issue found in the TAVI algorithm.
- Abbott ARCHITECT c16000 Processing Module, Class II – There is a potential to generate incorrect results on the instrument if particular error codes are not resolved before transitioning to the Running status.
- Elekta Unity, Image-Guided Radiation Therapy System, Class II – Users need to be aware when using these protocols for daily online plan adaptation that: 1) The images acquired using these protocols do not represent the average position of the anatomy during the respiratory motion cycle. The images are based on data acquired around full expiration. 2) The display of the images in the Elekta Unity Application software does not provide information about the protocol used to acquire the image eg. with or without respiratory triggering. Users must select an appropriate scan protocol that is representative of the respiratory phase used in the reference plan.
- Accelerate Pheno system, Class II – Rare isolates of Enterobacteriaceae may generate a susceptible meropenem Minimum Inhibitory Concentration (MIC) using the affected device and a resistant result by broth microdilution. In order to mitigate any risk associated with these rare isolates, the firm has implemented an Expert Rule that suppresses the meropenem result for isolates that display a profile consistent with those that generated a false-susceptible result. The firm has responded to this risk by implementing a set of suppression rules, in a software update, to prevent incorrect results.
- Vacuum Unit for CATSmart Vacuum Pump, Class II – The optional Vacuum Unit may stop working and display the failure message “Failure vacuum unit” during use.
- BD Synapsys Laboratory Solutions, Class II – BD Synapsys version 2.1 allowed the potential for a test order to be associated to an incorrect culture record.
- Omega Systems, Class II – The firm is recalling their Delta family of patient monitors software due to cybersecurity vulnerabilities, which may cause the device to reboot, lose functionality, and/or lose communication.
- Infinity Delta Family patient monitors, Class II – The firm is recalling their Delta family of patient monitors software due to cybersecurity vulnerabilities, which may cause the device to reboot, lose functionality, and/or lose communication.
- Monaco Radiation Treatment Planning (RTP) System, Class II – If Improve Target Dose was chosen as an optimization model in a previous treatment session, Monaco¿ will automatically use this optimization model again when proceeding with the online plan adaptation of a completion plan when it should not.
- BRANSIST Safire, Class II – Normal operation of the device is to power up the device in the morning, register the first patient, and then perform a fluoroscopy. If, however, the user powers up the device in the morning and makes an error by starting a fluoroscopy while the first patient is still being registered, the device application will abnormally terminate and require service intervention before it can be used again. This event will not occur after the first patient procedure.
- Sensis Vibe System, Class II – A software error may result in a system crash. The system must be restarted before the clinical procedure can be continued. The ablation treatment must be performed with a different system or without the use of the interface.
- TRINIAS for Diagnostic Imaging and Interventional Procedures, Class II – Two issues: Event 1: Normal operation of the device is to power up the device in the morning, register the first patient, and then perform a fluoroscopy. If, however, the user powers up the device in the morning and makes an error by starting a fluoroscopy while the first patient is still being registered, the device application will abnormally terminate and require service intervention before it can be used again. This event will not occur after the first patient procedure. Event 2: In rare cases a synchronization error may occur in the data transmission circuit in the device due to external noise. The visibility of the displayed image may be corrupted (images become bit-shifted abnormal images). If this event occurs, it may become difficult to see the object, which may hinder the examination and treatment.
- AMIA Automated Peritoneal Dialysis System, Class II – Potential for the software on Automated PD System cyclers which can cause shortened dwell times during a Cycle-Based therapy.
- Kaguya Automated Peritoneal Dialysis System, Class II – Potential for the software on Automated PD System cyclers which can cause shortened dwell times during a Cycle-Based therapy.
- Ingenuity TF PET/CT, Class II – A software update is being issued to correct multiple issues identified in the previous software version.
- Revolution CT systems with the SmartStep Option, Class II – On the Revolution CT systems equipped with the SmartStep Option, the Z location displayed on image annotation and on the gantry display may not be the actual location of the table and result in the system scanning at a location that is not what was confirmed by the operator.
- NeuViz 128 Multi-slice CT Scanner System, Class II – For surview scan length more than 500mm, if the user aborts or skips the reconstructed surview, the image may have the wrong field of view (FOV). When user plans and performs subsequent clinical scan with the wrong FOV the actual start and end position may not be consistent with the planned positions.
- Medtronic Carelink 2090 programmer, Class II – There is an error in how the programmer calculates and displays the remaining longevity value during a period of time (up to 2 years) prior to the device reaching its Recommended Replacement Time (RRT).
- GE Healthcare Centricity Universal Viewer Breast Imaging, Class II – When switching back & forth between multiple UV instances in the Windows taskbar, the patient images displayed on the mammo high resolution monitors may not show images of the patient selected on UV, which could result in an incorrect diagnosis.
- Atellica IM 1300 Analyzer, Class II – Multiple Issues Identified in Atellica Solution System Software.
- Eclipse Treatment Planning System with Proton Convolution Superposition algorithm, Class II – There is an anomaly with the Eclipse Treatment Planning System [TPS] Proton Convolution Superposition [PCS] dose calculation algorithm. The PCS algorithm calculates the water equivalent range incorrectly for non-square 3D CT images (either different number of pixels in X and Y, or non-square pixels). Before the dose is calculated, Eclipse resamples the CT images to create a calculation image with a maximum resolution of 256 x 256. The PCS algorithm assumes uniform resolution in X and Y directions, or that X = Y for all images, and erroneously sets the Y = X for dose calculation. For images that are not square, where either X<>Y, or the length of X does not equal the length of Y, the computed water equivalent range R is erroneous compared to the correct range R: 1) X=Y; R =R (R is correct); 2) XY R >R (R is too big).
- DX-D 600 Direct Radiography System, Class II – After an upgrade of the software of the Overhead Tube Crane, there were isolated cases in which the Overhead Tube Crane movement does not stop when the movement button is released. Instead of stopping while the movement button is released, the Tube Head Crane moves to the intended position. drops on the cobas e 801 module after a ProCell II M bottle changeover, which may lead to incorrect medical decisions with respect to diagnostics and patient treatment.
- FilmArray Gastrointestinal (GI) Panel, Class II – Elevated rates of false positive results for Campylobacter and Cryptosporidium have been identified.
- Brainlab RT Elements, Class II – There is a potential for an incorrect dose distribution calculation by Brainlab RT Elements software (for affected versions) under specific circumstances when using the Pencil Beam algorithm on the GPU (graphics card), as is the default system setting.
- KaVo Dental Technologies Cone-Beam CT System, Class II – Orthopantomograph OP 3D device has a defect in the device firmware versions 2.1.0 and 2.1.1.
- Innova IGS 630, Angiographic X-Ray, Class II – There is a potential for loss of the x-ray imaging function when the user changes field of view (FOV) from 30 cm to 20cm or from 20 cm to 30cm while releasing the fluoroscopy
footswitch pedal simultaneously. - Syngo Lab Data Manager System, Class II – Software Issues.
Health Canada Releases New Cybersecurity Guidance
Health Canada released the Pre-market Requirements for Medical Device Cybersecurity guidance on 17 June 2019. It became effective almost immediately (26 June 2019). This is a requirement for all products that consist of or contain software and are regulated as a medical device (Class I to Class IV) under the Medical Devices Regulations (the Regulations). This includes both in vitro diagnostic (IVD) and non in vitro diagnostic (nIVD) devices .
https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-healthproducts/medical-devices/application-information/guidancedocuments/cybersecurity-guidance.pdf
Health Canada Releases Draft SaMD Guidance
Health Canada released the draft software as a medical device (SaMD) guidance document on 23 January 2019. The guidance defines the following:
- SaMD is a medical device and includes in-vitro diagnostic (IVD) medical devices
- SaMD can run on general purpose (non-medical purpose) computing platforms, “without being part of” means software not necessary for a hardware medical device to achieve its intended medical purpose
- Software does not meet the definition of SaMD if its intended purpose is to drive a hardware medical device
- SaMD may be used in combination (e.g., as a module) with other products including medical devices
- SaMD may be interfaced with other medical devices, including hardware medical devices and other SaMD software, as well as general purpose software
- Mobile apps that meet the definition above are considered SaMD https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-healthproducts/public-involvement-consultations/medical-devices/software-medicaldevice-draft-guidance/software-medical-device-draft-guidance-eng.pdf
FDA Releases Requests for Feedback on the Q-Submission Program
The FDA issued a final guidance, Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program. Guidance for Industry and Food and Drug Administration Staff Document on May 7, 2019. The FDA’s Q-Submission Program provides submitters an opportunity to have early collaboration and discussions about medical device submissions.
https://www.fda.gov/media/114034/download
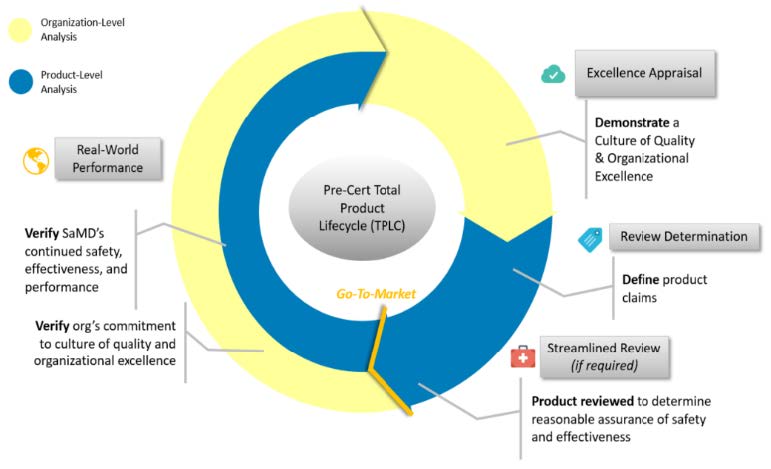
FDA Software Precertification (Pre-Cert) Pilot Program
The FDA has released in January 2019 the Software Precertification (Pre-Cert) Pilot Program, as outlined in the FDA’s Digital Health Innovation Action Plan. The plan will help inform the development of a future regulatory model that will provide more streamlined and efficient regulatory oversight of software based medical devices developed by manufacturers who have demonstrated a robust culture of quality and organizational excellence, and who are committed to monitoring real-world performance of their products once they
reach the U.S. market.
Proposed Key Components of a Future Pre-Cert Program:
FDA Software Pre-Cert Pilot Program Mid-Year Update
The FDA release the 2019 Mid-Year Update of the PreCert Program. The summary is that FDA will continue to test the program as outlined in the 2019 Test Plan, including testing the PreCert model on new SaMD submissions. The information collected will be used to determine whether the results of the Pre-Cert pathway align with the results of the traditional pathway and satisfy FDA’s regulatory requirements for safety and effectiveness. The update can be found in the following link.
https://www.fda.gov/media/129047/download
FDA Software Pre-Cert Pilot Program Article
Article from MobileHealthNews 8/2/19 – How many companies stand to benefit from the FDA Pre-Cert program? Fewer than you might think. The author is pessimistic. This will give you another point of view on this program.
https://www.mobihealthnews.com/content/how-many-companies-standbenefit-fda-pre-cert-program-fewer-you-might-think
FDA Medical Device Safety Action Plan
The FDA issued the Medical Device Safety Action Plan: Protecting Patients, promoting Public Health on January 22, 2019. The FDA wants medical devices to have mandatory built-in update mechanisms and plans to ask Congress for
more funding and regulatory powers to improve its approach towards medical device safety, including on the cybersecurity front.
The FDA document reveals several of the FDA’s plans, including the desire to force device makers to include mandatory update systems inside products for the purpose of delivering critical security patches.
https://www.fda.gov/media/112497/download
FDA Draft Guidance Content of Premarket Submissions for Management of Cybersecurity in Medical Devices
The FDA Issues draft Guidance Content of Premarket Submissions for Management of Cybersecurity in Medical Devices on October 18, 2018. The draft guidance relates to having the manufacturer prepare and submit the Cybersecurity Bill of Materials (CBOM), which includes a list that includes but is not limited to commercial, open source, and off-the-shelf software and hardware components that are or could become susceptible to vulnerabilities. Hospitals, healthcare units, contractors and users will be able to consult the medical device’s CBOM and determine how it functions, what software is needed for what feature, and what technologies are used in each device.
The idea is to help device owners “better manage their networked assets and be aware of which devices in their inventory or use may be subject to vulnerabilities.”
https://www.fda.gov/media/119933/download
There have been numerous articles written since the draft guidance release. Here are a few of them with their links:
Meddevice: “Cybersecurity Bill of Materials for Medical Devices: What’s Next?”
https://www.meddeviceonline.com/doc/cybersecurity-bill-of-materials-formedical-devices-what-s-next-0001
Alienvault: “Software Bill of Materials (SBoM) – Does It Work for DevSecOps?”
https://www.alienvault.com/blogs/security-essentials/software-bill-ofmaterials-sbom-does-it-work-for-devsecops
ANSI/AAMI/UL 2800-1:2019 – Standard for Safety for Medical Device Interoperability
This standard specifies a baseline set of requirements for assuring safe and secure interoperability for Interoperable Medical Systems. The standard employs a life cycle process approach to organizing requirements, providing a set of interoperability planning, realization,
deployment, and monitoring activities that incorporate cross-cutting requirements for security and risk management. AAMI/UL 2800-1 also provides supplementary guidance on key clinical and engineering properties essential for ensuring effective interoperability.
FDA’s Software Related Prioritized Medical Device Guidance Documents to Publish in FY 2019 (“A-list”)
Final Guidance Topics:
- The Least Burdensome Provisions: Concept and Principles Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act
- Clinical and Patient Decision Support Software
- Multiple Function Device Products: Policy and Considerations Draft Guidance Topics:
- Content of Premarket Submissions for Cybersecurity of Medical Devices of Moderate and Major Level of Concern
- Computer Software Assurance for Manufacturing, Operations, and Quality System Software
- Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
It’s imperative to see the direction that the FDA is going with software regulation. The emphasis will be on handling decision support software (until now this type of software has mainly flown under the radar), upgrades in cybersecurity requirements and software used in the organization (bring the FDA in line with ISO 13485:2016).
The additional issue is the reorganization of the Content of Premarket Submissions for Software Contained in Medical Devices, which determines the software’s Level of Concern (LOC) and the corresponding documentation required to submit. I haven’t been able to get any inputs yet on the new guidance, so if there is something out there, I would love to get a peek.
FDA Recognizes Defect Taxonomy Consensus Standard
The FDA recognized the ANSI AAMI SW91:2018 Classification of defects in health software as a consensus standard. The FDA recognition statement for this standard does not indicate any specific use in premarket submissions or relevant FDA guidances. It simply states it supports existing policies. This standard is lengthy and technical in terms of its approach to defect classification and focuses on technical and process root causes rather than clinical safety and effectiveness impact. One possible use of this standard is to consider some of the types of defects including those in the Annexes when doing software risk analysis and root cause investigations. It is possible in the future some FDA staff may familiarize themselves with this standard and ask if some of these types of defects have been considered during development, failure analysis, or in defect trending for handling CAPAs.
FDA Draft Guidance on Technical Performance Assessment of Quantitative Imaging in Device Premarket Submissions
The FDA released a draft guidance providing manufacturers and FDA staff with detailed recommendations on assessing the technical performance of quantitative imaging devices and how the documentation from those assessments should be provided in premarket submissions in April 2019. From a big picture perspective, one should remember the overall goal is to “provide performance specifications for the quantitative imaging functions, supporting performance data to demonstrate that the quantitative imaging functions meet those performance specifications, and sufficient information for the end user to obtain, understand, and interpret the values provided by the quantitative imaging functions.”
https://www.fda.gov/media/123271/download
FDA Medical Device Development Tool (MDDT) Qualification
The FDA has provided guidance on the methods and approaches to qualify a medical device development tool so that medical device manufacturers or sponsors can use them to support the development and evaluation of medical devices. The manufacturer is expected to ensure the tool produces “scientifically-plausible measurements” and works as intended within the specified intended use of the tool. Some specific points in the guidance for medical device development tool qualification are:
Specification of the intended use and the scope of the product areas covered What the medical device development tool measures or produces as an output Role of the medical device development tool with respect to clinical uses or study parameters Phases of the medical device development lifecycle where the medical device development tool will be used https://www.fda.gov/medical-devices/science-and-research-medicaldevices/medical-device-development-tools-mddt.
Proposed Regulatory Framework for Modifications to Artificial
Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback
FDA Regulation of Artificial Intelligence (AI) and Machine Learning in Software as a Medical Device On April 2, 2019, the FDA published a discussion paper “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback” that describes the FDA’s foundation for a potential approach to premarket review for artificial intelligence and machine learning-driven software modifications.
FDA’s current software framework would be likely to require premarket submissions for most changes to AI based devices. In the new framework, the FDA introduces a “predetermined change control plan” in premarket submissions. This plan would include the types of anticipated modifications— referred to as the “Software as a Medical Device Pre-Specifications”—and the associated methodology being used to implement those changes in a controlled manner that manages risks to patients —referred to as the
“Algorithm Change Protocol.”
https://www.fda.gov/media/122535/download
Proposed IEC 62304 Edition 2, Health Software – Software Life Cycle Processes
The proposed IEC 62304 edition 2 standard has been rejected again. The standard went back to committee and they need to issue an update for review.
Software Safety Classes (IEC 62304) versus Levels of Concern (FDA)
Both, IEC 62304 and the FDA (Content of Premarket Submissions for Software Contained in Medical Devices) distinguish three different categories of medical device software. The IEC 62304 uses the software safety classes (SSC) and the FDA guideline uses the Level of Concern (LOC). This causes much confusion.
- The SSC is defined as follows in IEC 62304:2006 + A1:2015:
The software system is software safety class A if:
– the software system cannot contribute to a hazardous situation;
or
– the software system can contribute to a hazardous situation which does not result in unacceptable risk after consideration of
risk control measures external to the software system. - The software system is software safety class B if:
– the software system can contribute to a hazardous situation which results in unacceptable risk after consideration of risk
control measures external to the software system and the resulting possible harm is non-serious injury. - The software system is software safety class C if:
– the software system can contribute to a hazardous situation which results in unacceptable risk after consideration of risk control measures external to the software system and the resulting possible harm is death or serious injury.
The LOC is determined as follows in the FDA’s Content of Premarket
Submissions for Software Contained in Medical Devices:
- Major: We believe the level of concern is Major if a failure or latent flaw could directly result in death or serious injury to the patient or operator. The level of concern is also Major if a failure or latent flaw could indirectly result in death or serious injury of the patient or operator through incorrect or delayed information or through the action of a care provider.
- Moderate: We believe the level of concern is Moderate if a failure or latent design flaw could directly result in minor injury to the patient or operator. The level of concern is also Moderate if a failure or latent flaw could indirectly result in minor injury to the patient or operator through incorrect or delayed information or through the action of a care provider.
- Minor: We believe the level of concern is Minor if failures or latent design flaws are unlikely to cause any injury to the patient or operator. The SSC classes determine the software life-cycle development processes to be performed and documented. Class A has the least processes and documentation required and Class C has the most. The SSC is determined at the beginning in the project.
The LOC determines the document to be submitted as part of the submission (and not as part of the development process). The LOC must be determined before the submission. It has been known in numerous cases, that the FDA has determined the LOC is different than what the company determined (the FDA always wins). There is a virtual connection between the SSC and the LOC, but they both relate to different aspects (processes and documentation vs. documentation to be submitted) and should be handled accordingly.
FDA Responses to 510K Submissions – Software
We are still receiving responses from the FDA concerning their software. This means that this is becoming the state of the practice for the FDA. These responses relate to the run-time testing, and cybersecurity. Below is shown the wording received from the FDA in all the cases:
- The submission did not include information on the tools, such as static analysis tools, that you used to detect run-time errors. This information is needed to assess whether good coding practices have been implemented to prevent common coding errors which may adversely affect the safety of the device. Please provide this information. For any such tool used, please identify what error types the tool detects, your method and process of applying the tool(s), and a summary report and/or conclusion about the results. Note: some common run-time errors are:
a. Un-initialized variables
b. Type mismatches
c. Memory leaks
d. Buffer over/under flow
e. Dead and unreachable code
f. Memory/heap corruption
g. Unexpected termination
h. Non-terminating loops
i. Dangerous Functions Cast
j. Illegal manipulation of pointers
k. Division by zero
l. Race conditions
- The information security and cybersecurity of the device is needed to evaluate the cybersecurity risks and the associated controls. The FDA has been asking for the cybersecurity even from devices that have no connectivity.
a. Please discuss in detail, information on your design considerations, including mitigations pertaining to intentional and unintentional
cybersecurity risks including:
b. A specific list of all cybersecurity risks that were considered in your design.
c. A specific list and justification for all cybersecurity controls that you established, and the justification as to why such controls are
adequate. Please provide the evidence that the controls perform as intended.
d. Please ensure that you address information confidentiality, integrity and availability.
e. Please incorporate, as appropriate, the information identified here in your Hazard Analysis.
3. The FDA has been reading the software documentation, including the Risk Analysis, SRS, SDD, STD, STR, Traceability Report, OTS Report, Cybersecurity, etc. They have been raising issues as shown in the following:
- SRS: contradictions and not containing information necessary to understand the requirements for your device software; requirements
related to programming language requirements or to the interfaces. - SDD: high-level architecture and does not include the level of detail expected for software architecture; does not include information
necessary to ensure that your software is safe and effective for the intended use of the device; missing information for all the third-party devices used by your system. - Traceability Report: traceability documentation does not link between requirements to the hazards
- Testing: it doesn’t include a summary of the static analysis, examples of unit integration testing, and a summary of the results.
We are highly recommending to clients several remediations:
- SSC Class B/Moderate LOC – software require tools to test the software for run-time errors. We are recommending using static code analysis tools. There are low end tools that should be used, e.g., Source Code Analysis package for medical device companies from Parasoft (C/C++, Java, C#/VB.NET), Microsoft Visual Studio Static Code Analysis (C/C++), IAR C-STAT static analysis (C/C++), etc.
- SSC Class C/Major LOC/Special Guidance/PMA – FDA will ask for a SCA report. We highly recommend using one of the tools that we know the FDA has evaluated. A partial list of these tools is Parasoft, Coverity, Polyspace, PQRA, Klocwork, Grammatech and LDRA.
- A cybersecurity report should be prepared for submission to the FDA based upon the threat analysis.
- Using tools for cybersecurity testing, penetration testing, etc. When choosing SCA and cybersecurity tools, check the local support. Even though everyone offers Internet support, nothing beats having the support done locally by someone who has the experience and speaks your language.
Summary
There are many ways to screw up your software in the medical device. It doesn’t take too much talent to do this and companies are doing it daily. Many companies mess up royally and don’t know how to get out of the mess. In many cases, they don’t even know that they are in deep trouble. You can work properly without breaking the bank. There are many ways to handle the software development/maintenance life cycle and the software validation.
If there are any questions or requests, please feel free to contact us.
Mike
Related Posts


